


































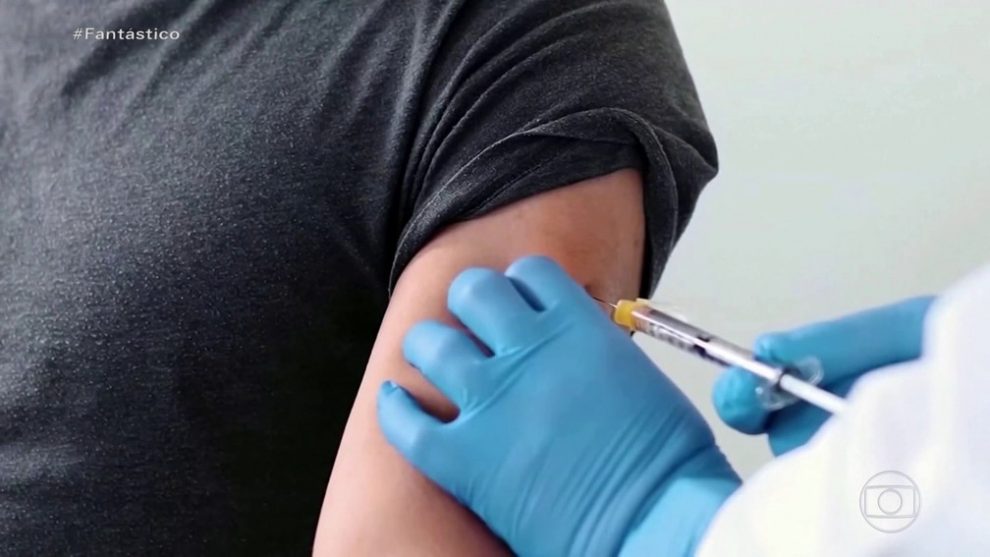
Diretora da Anvisa disse que agência ainda não recebeu nenhum pedido de uso emergencial nem pedido de registro de vacinas. No dia 2 de dezembro, a agência já tinha definido os requisitos para o pedido.
A Diretoria Colegiada da Anvisa (Agência Nacional de Vigilância Sanitária) aprovou nesta quinta-feira (10) as regras para a autorização temporária de uso emergencial, em caráter experimental, de vacinas contra a Covid-19 (veja principais pontos abaixo).
A decisão foi unânime entre os diretores da agência. No dia 2 de dezembro, a Anvisa já tinha definido os requisitos para o pedido de uso emergencial da vacina.
Na prática, a medida abre caminho para que empresas possam fazer esse pedido de emergência.
“A autorização de uso emergencial é um mecanismo que pode facilitar a disponibilização e o uso das vacinas contra Covid-19, ainda que não tenham sido avaliadas sob o crivo do registro, desde que cumpram com os requisitos mínimos de segurança, qualidade e eficácia”, disse Alessandra Bastos Soares, diretora da Anvisa.
A agência ainda não recebeu nenhum pedido de uso emergencial nem pedido de registro de vacinas. Soares ressaltou que esse pedido deve ser feito pela empresa.
“Qualquer autorização concedida pela Anvisa, qualquer anuência, só será feita diante de um pleito. A vacina só terá autorização de uso emergencial e experimental se houver o pleito realizado por alguma empresa”, disse Soares.
A Anvisa ressalta que poderá modificar, suspender ou cancelar a autorização temporária a qualquer momento, com base em elementos técnicos e científicos.
A concessão estabelecida pela Anvisa segue o modelo de autorizações emergenciais adotadas em outros países, como Reino Unido, Estados Unidos e Canadá, e vale apenas para o período de pandemia e até a vacina receber o registro definitivo.
Quatro vacinas estão em testes de fase 3 no Brasil: a da Pfizer, a de Oxford, a da Johnson e a da Sinovac
Principais pontos do uso emergencial
- Cada pedido deve ser feito pela empresa desenvolvedora e será analisado de forma independente;
- Decisão será tomada pela Diretoria Colegiada da Anvisa;
- Serão considerados estudos não-clínicos e clínicos (em humanos);
- Serão itens avaliados: qualidade, boas práticas de fabricação, estratégias de monitoramento e controle, e resultados provisórios de ensaios clínicos;
- Empresa interessada deverá comprovar que a fabricação e a estabilidade do produto garantem a qualidade da vacina;
- Estudo clínico na fase 3 – última etapa de testes – deve estar em andamento e conduzido também no Brasil;
- Vacina com uso emergencial liberado não pode ser comercializada, ela só pode ser distribuída no sistema público de saúde;
- Liberação de uso emergencial pode ser revogada pela Anvisa a qualquer momento.
‘Lei Covid’
A utilização de uma vacina contra a Covid-19 no Brasil depende da aprovação pela Anvisa. A permissão pode ser conseguida, basicamente, por dois caminhos.
O primeiro está diretamente ligado aos dois tipos de registro (tradicional ou emergencial) que podem ser dados pela Anvisa.
Já a segunda possibilidade é baseada na chamada “Lei Covid”, que libera o uso se o imunizante tiver aval expedido por uma agência do exterior, independentemente de registro pela Anvisa.
- Anvisa – registro definitivo: os desenvolvedores submetem o pedido de registro à Anvisa apenas após concluírem as 3 fases de testes da vacina. Para acelerar o trâmite, a agência criou o procedimento de submissão contínua de dados.
- Anvisa – uso emergencial: permite aos desenvolvedores enviarem os dados que comprovem eficácia e segurança antes de terminarem a fase 3 da vacina;
- Lei Covid – Prevê que a Anvisa terá o prazo de 72 horas para conceder a autorização caso o imunizante tenha conseguido registro no Japão, nos EUA, na Europa ou na China. Caso o prazo não seja cumprido e a Anvisa não se manifeste, a autorização é concedida automaticamente.
Por Mariana Garcia, G1





































Adicionar comentário